
Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing
Pös, O.1,2, Budis, J.3,4,5, Kubiritova, Z.6,7, Kucharik, M.8, Duris, F.9,10, Radvanszky, J.11,12, Szemes, T.13,14,15
- 1Faculty of Natural Sciences, Comenius University, 841 04 Bratislava, Slovakia
- 2Geneton Ltd., 841 04 Bratislava, Slovakia
- 3Geneton Ltd., 841 04 Bratislava, Slovakia
- 4Comenius University Science Park, 841 04 Bratislava, Slovakia
- 5Slovak Center of Scientific and Technical Information, 811 04 Bratislava, Slovakia
- 6Faculty of Natural Sciences, Comenius University, 841 04 Bratislava, Slovakia
- 7Institute for Clinical and Translational Research, Biomedical Research Center, Slovak Academy of Sciences, 845 05 Bratislava, Slovakia
- 8Geneton Ltd., 841 04 Bratislava, Slovakia
- 9Geneton Ltd., 841 04 Bratislava, Slovakia
- 10Slovak Center of Scientific and Technical Information, 811 04 Bratislava, Slovakia
- 11Faculty of Natural Sciences, Comenius University, 841 04 Bratislava, Slovakia
- 12Institute for Clinical and Translational Research, Biomedical Research Center, Slovak Academy of Sciences, 845 05 Bratislava, Slovakia
- 13Faculty of Natural Sciences, Comenius University, 841 04 Bratislava, Slovakia
- 14Geneton Ltd., 841 04 Bratislava, Slovakia
- 15Comenius University Science Park, 841 04 Bratislava, Slovakia
Abstract
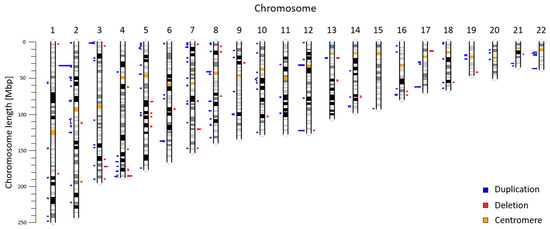
Copy number variants (CNVs) are an important type of human genome variation, which play a significant role in evolution contribute to population diversity and human genetic diseases. In recent years, next generation sequencing has become a valuable tool for clinical diagnostics and to provide sensitive and accurate approaches for detecting CNVs. In our previous work, we described a non-invasive prenatal test (NIPT) based on low-coverage massively parallel whole-genome sequencing of total plasma DNA for detection of CNV aberrations ≥600 kbp. We reanalyzed NIPT genomic data from 5018 patients to evaluate CNV aberrations in the Slovak population. Our analysis of autosomal chromosomes identified 225 maternal CNVs (47 deletions; 178 duplications) ranging from 600 to 7820 kbp. According to the ClinVar database, 137 CNVs (60.89%) were fully overlapping with previously annotated variants, 66 CNVs (29.33%) were in partial overlap, and 22 CNVs (9.78%) did not overlap with any previously described variant. Identified variants were further classified with the AnnotSV method. In summary, we identified 129 likely benign variants, 13 variants of uncertain significance, and 83 likely pathogenic variants. In this study, we use NIPT as a valuable source of population specific data. Our results suggest the utility of genomic data from commercial CNV analysis test as background for a population study.